
References
Understanding the pathogenesis of heart failure

Abstract
Heart failure is a complex clinical syndrome. Jamshid Easa, Jacob Chappell and David Warriner explain the pathogenesis behind the most common form of this condition
Heart failure a complex clinical syndrome due to impairment of ventricular filling or contraction, resulting in a constellation of physical symptoms and signs, primarily due to salt and water retention. In this clinically focused review of the pathophysiology, we will summarise the main consequences of the most common type, left ventricular failure; knowledge of which is essential for those working in general practice due to the high prevalence of heart failure in the community and to aid understanding of the various pharmacotherapies that work to act on the pathological mechanisms.
Heart failure is defined as a ‘complex clinical syndrome that results from any structural or functional impairment of ventricular filling or ejection of blood’ (Dickstein et al, 2008). Patients generally present with a constellation of physical symptoms, such as fatigue and dyspnoea, and signs, such as peripheral and pulmonary oedema. It can be categorised in many ways, but the most common is left or right ventricular failure, with left ventricular failure being further subdivided into either systolic dysfunction (impaired ventricular contraction and ejection) or diastolic dysfunction (impaired ventricular relaxation and filling) (Figure 1). For the purposes of this article, we shall focus mainly on the most common, left ventricular failure (LVF), also known as left ventricular systolic dysfunction – heart failure or LVSD-HF for short (unless otherwise stated).
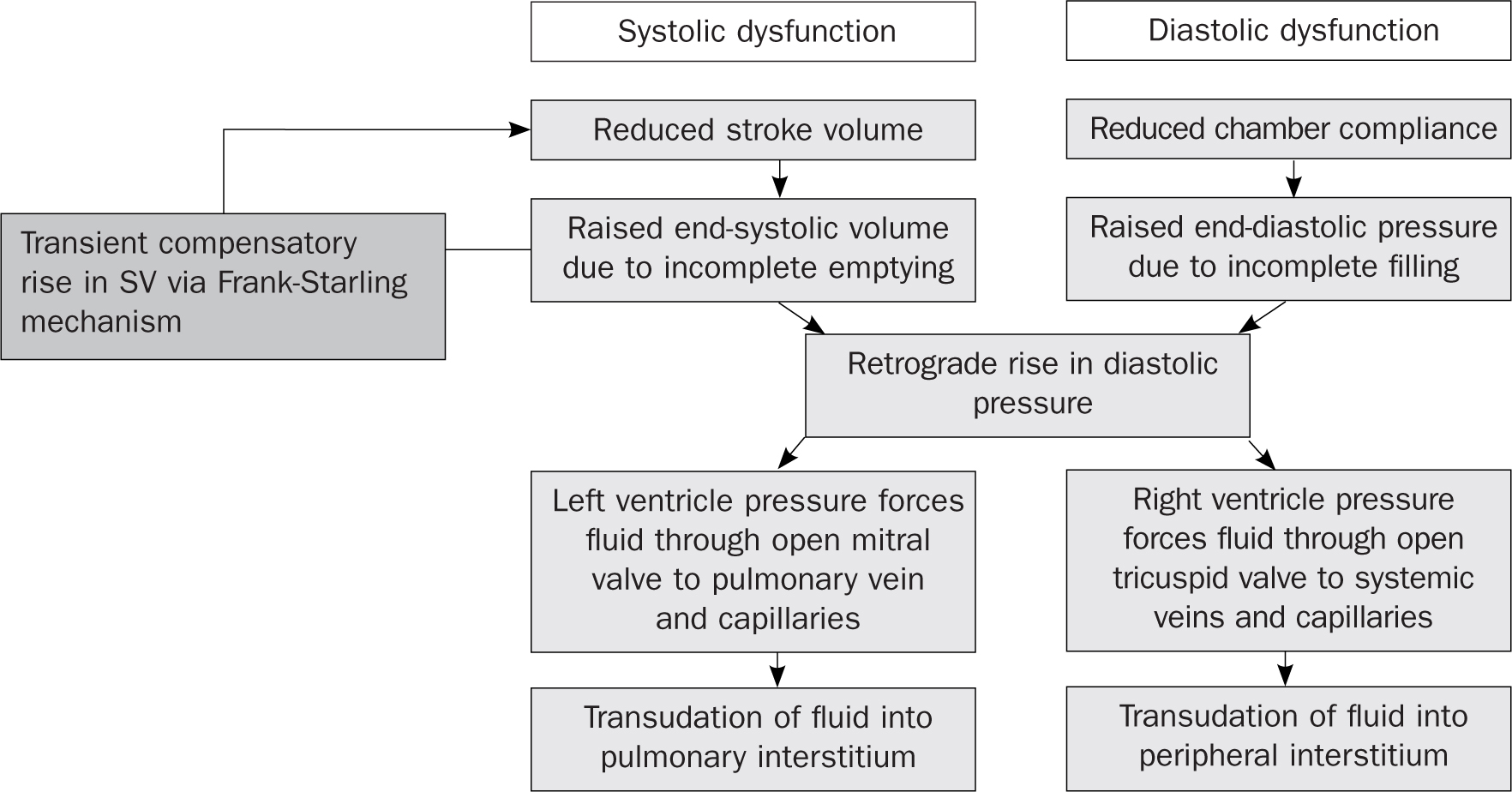 Figure 1. Mechanisms of cardiogenic pulmonary and peripheral oedema in left ventricular failure
Figure 1. Mechanisms of cardiogenic pulmonary and peripheral oedema in left ventricular failure
Register now to continue reading
Thank you for visiting Practice Nursing and reading some of our peer-reviewed resources for general practice nurses. To read more, please register today. You’ll enjoy the following great benefits:
What's included
-
Limited access to clinical or professional articles
-
New content and clinical newsletter updates each month